Среди наследственных мутаций примерно в одном из 5000 случаев встречается заболевание аутосомно-доминантного типа наследования под названием «синдром Марфана». Что это за патология, и почему она интересует ученых и врачей, занимающихся проблемами костно-мышечной системы человека?
- Марфана синдром — что это такое?
- Что означает аутосомно-доминантный тип наследования
- По каким симптомам определяется синдром Марфана
- Конституция при синдроме Марфана
- Ортопедические заболевания при синдроме Марфана
- Поражения сердечно-сосудистой системы
- Как синдром Марфана отражается на зрении
- Кожные, вегетативные и трофические расстройства
- Другие патологии
- Синдром Марфана у детей
- А как с интеллектом у больных с синдромом Марфана?
- Диагностика синдрома Марфана
- Лечение синдрома Марфана
- Профилактика осложнений
Марфана синдром — что это такое?
Генетическая суть синдрома Марфана заключается в изменении фрагмента кода двухкомпонентного белка фибриллина-1, приводящая к патологиям соединительных тканей:

- в них начинают накапливаться гликозаминогликаны (по типу хондроитина и гиалуроновой кислоты), и они чрезмерно растягиваются;
- нарушается обмен аминокислот, входящих в состав коллагена;
- появляется недостаточность функции коры надпочечников;
- в моче наблюдается избыток мукополисахаридов и оксипролина.
Соединительные ткани входят в состав как КМС, так и всех органов человека, поэтому любые патологические изменения в них способны привести как к ортопедическим заболеваниям, так и болезням многих органов и систем.
Что означает аутосомно-доминантный тип наследования
Аутосомно-доминантное наследование во много раз повышает вероятность проявления генетической мутации у потомства.
Каждая хромосома состоит из множества генов, которые несут как доминантные признаки (аллели), так и рецессивные (они подавляются при соединении доминантного и рецессивного алелля).
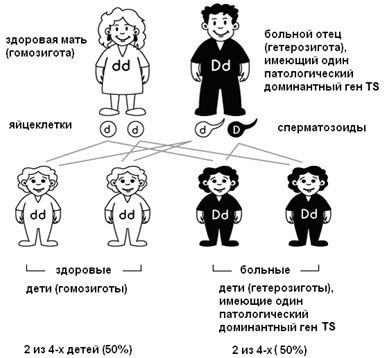
При скрещивании двух родительских гамет, содержащих хотя бы один дефективный доминантный аллель (пусть это будет ген болезни Марфана), возможно появление четырех новых зигот, две из которых содержат только одинаковые рецессивные, в нашем случае, признаки (отсутствие синдрома Марфана) — они называются гомозиготы, а две — и доминантный, и рецессивный (они называется гетерозиготами).
Из гомозиготы со «здоровыми» одинаковыми аллелями однозначно разовьется здоровый ребенок. Генотип гетерозиготы, где доминирует аллель, несущий синдром Марфана, определяет будущий нездоровый фенотип с симптомами этого заболевания.
Получается, что всего на всего один дефективный, но доминантный родительский ген наследует половина потомства в первом поколении (если хотя бы один родитель болен). Но доминирующий признак способен передаваться дальше по наследству в следующих поколениях даже от здоровых «гомозиготных» детей, у которых оба гена рецессивные. Вероятность наследования мутации от человека со здоровым внешним фенотипом, но в генотипе которого имеется ген болезни, в первом поколении составляет ¼, в третьем — ⅛
и т. д.
При этом пройдя даже через тысячи поколений, доминантный ген никуда не исчезнет и никогда не утратит своего свойства подавлять рецессивные гены. Пускай вероятность встречи двух гамет, одна из которых содержит доминантный ген с дефектом, будет расценена как 1/1000, но если она случайно произойдет, то новорожденный неизбежно приобретает доминантный признак, то есть наследственное заболевание в своем фенотипе.
При аутосомно-рецессивном типе вероятность наследования значительно снижается: есть шансы рождения больного ребенка, когда оба родителя больны, или оба носят рецессивные признаки в наследственном генотипе.
Эту особенность наследования рецессивных и доминантных признаков в гомозиготах и гетерозиготах впервые установил биолог-генетик Мендель, при селекции разных сортов гороха.
Особенностью всех наследственных заболеваний является их неизлечимость, так как человечество пока не научилось изменять генетический код.
По каким симптомам определяется синдром Марфана
Для с. М свойственна классическая триада симптомов:
- нарушения в ОДС;
- сердечно-сосудистые патологии;
- глазные заболевания (могут быть врожденными).
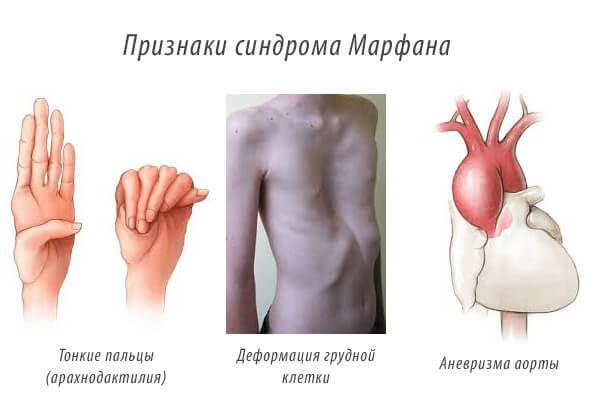
Помимо этого, заболеванию свойственные кожные и трофические изменения. Синдром может проявляться явно, захватывая много диагностических признаков, и давать скупой фенотип.
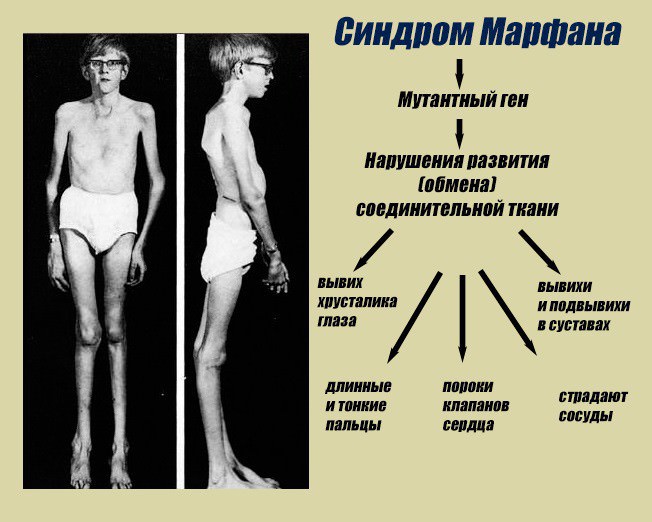
Конституция при синдроме Марфана
Больные с наследуемым синдромом Марфана отличаются выраженными скелетными диспропорциями, деформациями, дистрофией:
- высокий рост, длинные руки и ноги и короткое туловище;
- кисти и стопы удлиненные, в основном из-за арахнодактилии (паукообразных пальце);
- череп вытянутой формы, скулы узкие (признаки долихостеномелии);
- челюсть выступает;
- глаза близко и глубоко посажены (птичий тип);
- зубы растут неровно и скученно;
- небо высокое (готическое);
- подкожного жира мало;
- грудная клетка вдавлена либо килевидной формы;
- между тонкими длинными ребрами большой промежуток;
- уменьшение эпигастрального угла.
На фото: Больной синдромом Марфана
Ортопедические заболевания при синдроме Марфана
При данной врожденной патологии наблюдается:
- Гипермобильность суставов из-за повышенной растяжимости связок, из-за чего люди с синдромом Марфана подвержены частым подвывихам и вывихам.
- Позвоночные деформации: сколиоз, выраженные сутулость и поясничный лордоз.
- Дисплазии тазобедренного сустава, в частности недоразвитие вертлужной впадины, что приводит к раннему коксартрозу.
- Плоскостопие.
- Деформация и развитие контрактур на пальцах и локтях.
- Остеопороз, проявляемый в разреженной неоднородной структуре костных балок, известковых отложениях, пучков коллагеновых волокон. Осложнения в виде частых патологических переломов.
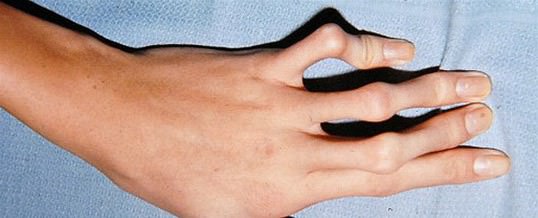
Поражения сердечно-сосудистой системы
Большой опасности при синдроме Марфана подвергаются важные элементы сердечно-сосудистой системы — аорта, миокард, легочные стволы. Из-за разрыхления эндотелия, волокна соединительных тканей разрыхляются, разбухают; происходит сужение просвета сосудов, миксоматозное утолщение стен клапана миокарда.
Кардиально-сосудистая диагностика устанавливает:
- слабо развитый каркас легочного ствола и аорты;
- расширение (дилатацию) корня аорты;
- расширение границ сердца;
- наличие систолических и диастолических шумов;
- повышенную нагрузку на правые желудочки.
Последствия всех этих пагубных деструктивных изменений соединительных тканей:
- Пролапс митрального клапана (происходит в большинстве случаев).
- Инфекционный эндокардит.
- Сердечная недостаточность.
- Расслаивающаяся аневризма аорты и легочного ствола.
- Пневмоторакс.
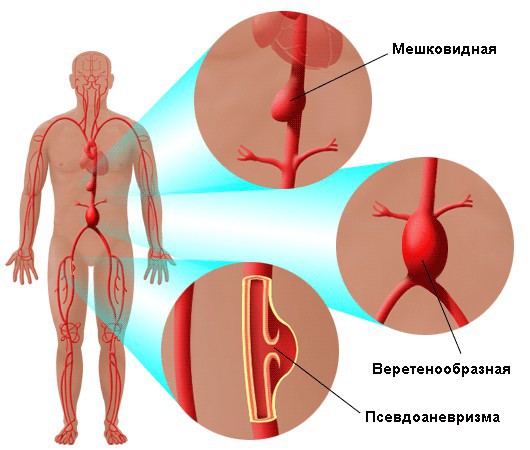
Расслоение стенок сосудов часто приводит к их разрыву и ранней смерти пациента. Без поддерживающей терапии больные с явной сердечно-сосудистой симптоматикой редко доживают до 50 лет, впрочем, имеются случаи и продолжительной жизни людей с подобным диагнозом.
Как синдром Марфана отражается на зрении
Слабость связок пагубно сказывается не только на состоянии ОДС, но и на зрении.
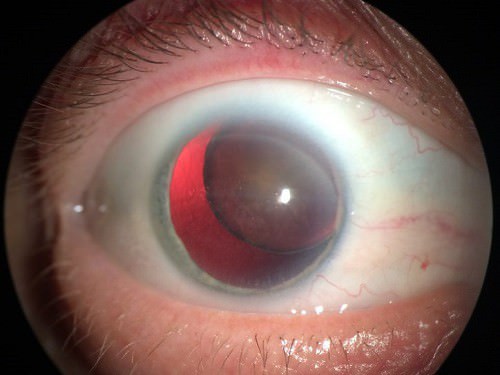
У половины больных наблюдается:
- полное или частичное смещение хрусталика (эктопия);
- близорукость (миопия);
- возникает риск отслоения сетчатки;
- при переднем вывихе хрусталика может повредиться роговица глаза, образоваться воспаление (увеит) или глаукома.
Кожные, вегетативные и трофические расстройства
Из-за чрезмерного растяжения кожи на ней часто образуются растяжки (стрии) в грудной, плечевой и поясничных областях.
Вегетативно-трофические признаки:
- сильное потоотделение;
- холодные ступни и кисти;
- вялость;
- мертвенная бледность кожи с синюшным оттенком (акроцианоз);
- мраморный ладонный рисунок.
Такое состояние кожи может быть и при отсутствии пролапса митрального клапана.
Другие патологии
У больных синдромом Марфана часто возникает сужение пояснично-крестцового канала, что проявляется:
- в симптомах острой радикулопатии — боли в пояснице, отдающей в одну или обе конечности;
- периодической (перемежающейся) хромоте;
- слабости и снижения рефлексов в ногах.
Также из-за сосудистых патологий при синдроме Марфана высока опасность инсульта (кровоизлияния в мозг) и кровотечений в субарахноидальном пространстве.
Нередко у людей, страдающих с. Марфана развивается почечная недостаточность, образуются кисты в почках и печени.
Синдром Марфана у детей
Определяется в первую очередь по анатомическим особенностям, перечисленным выше. Больные дети отличаются астеническим конституционным типом. У них могут ранние проблемы со зрением, они часто болеют ОРВИ и гриппом, пневмонией.
Клинические признаки сердечно-сосудистых патологий могут быть слабо выражены у детей. Постепенно они прогрессируют и проявляются по мере взросления в виде тахикардии, аритмии, сердечной и дыхательной недостаточности.
А как с интеллектом у больных с синдромом Марфана?
Интеллект у больных с. М. обычно не страдает. Существует даже предположение, что наличие синдрома Марфана способствует развитию гениальных способностей. Из известных знаменитостей этой наследственной болезнью страдали сказочник Ганс Кристиан Андерсен, 16-й американский президент Авраам Линкольн, великие композиторы Паганини и Рахманинов, некоторые современные актеры и музыканты.

Диагностика синдрома Марфана
Для установления правильного диагноза проводят клинический осмотр, составляют семейный анамнез. Оценку состояния производят по стандартным таблицам, включающим большие и малые критерии.
Проводят инструментальное обследование:
- рентгенографию позвоночника и грудной клетки;
- ЭКГ;
- УЗДС сосудов;
- УЗИ сердца;
- аортографию;
- ангиографию;
- офтальмологическую и неврологическую диагностику.
Лабораторное обследование включает:
- биохимические анализы крови и мочи;
- молекулярно-генетическое исследование с целью обнаружения мутировавшего гена.
ДНК — анализ является окончательным верификационным методом диагностики синдрома Марфана.
Лечение синдрома Марфана
Лечение патологии направлено на смягчение симптомов и продление жизни.

Это:
- Поддержание необходимого уровня гормонов, вырабатываемых корой надпочечников: из-за недостаточности функций надпочечников назначаются препараты ГКС.
- Профилактика аневризмы аорты при помощи бета-адреноблокаторов.
- При сильно расширенном корне аорты проводится коррекционная операция.
- При эктопии хрусталика производится лазерная офтальмологическая операция, при миопии выписываются соответствующие минусу зрения очки.
Торакопластика при деформациях грудной клетки не рекомендуется из-за угрозы осложнения: расслоения аневризмы А. и ее последующего разрыва.
Профилактика осложнений
Больные должны:
- избегать всю жизнь больших нагрузок и травм;
- периодически проходить обследования у ортопеда, кардиолога, офтальмолога, невролога и других врачей;
- заботиться о своих сосудах, принимая ангиопротекторы;
- соблюдать диету № 10, назначаемую при сердечно-сосудистых заболеваниях.
Женщинам, страдающим данным недугом, лучше не планировать беременность.
Соблюдение этих правил позволяет существенно увеличить продолжительность жизни у больных синдромом Марфана, хотя и не приводит к излечению от нее.