Спинальная амиотрофия (спинальная мышечная атрофия или СМА) — это неизлечимое, почти всегда наследуемое заболевание, вызванное мутацией гена в 5-й хромосоме.
Мутация гена СМА приводит к недостатку белка, который необходим для построения белковых РНК-структур, и к недостаточному развитию поперечнополосатых мышц, преимущественно нижних конечностей, а также шейного отдела и головы.
Болезнь может проявиться с момента рождения, и даже когда плод еще в утробе, и в любой период жизни. У новорожденных спинальная амиотрофия чаще всего приводит к ранней смерти, но в некоторых случаях она может иметь мягкие формы, в основном в пожилом возрасте. Рассмотрим подробнее особенности этой патологии.

Спинальная амиотрофия — все о болезни
История и статистика
СМА — достаточно редкое заболевание, открытое немецким врачом Верднигом в 1891 г. Заболевает ею один человек из 6 — 10 тыс., однако носителем рецессивного гена СМА является каждый 50-й человек.
В 1898 г. Вердниг и еще один ученый Гофман установили, что причиной СМА является дегенеративное поражение и недостаточное количество двигательных (моторных) нейронов передних рогов спинного мозга — SMN (survival motor neurons).
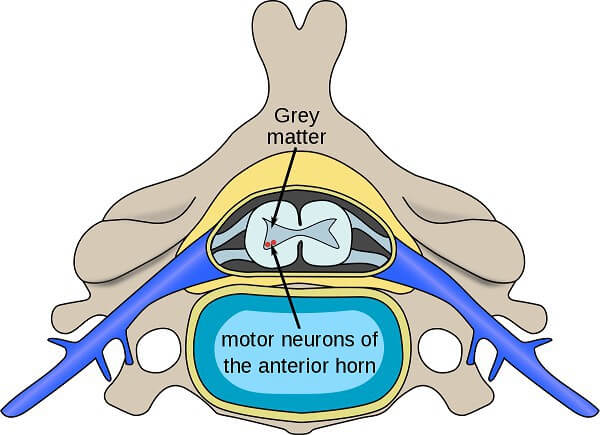
Уже в 20-м веке (в 1956 г.) другие ученые Кугельберг и Веландер открыли менее злокачественную, с более мягкими проявлениями, форму СМА, которой болеют в ювенильном и взрослом возрасте.
Тип наследования при спинальной амиотрофии
Болезнь может наследоваться по любому из типов:
- аутосомно-доминантному;
- аутосомно-рецессивному;
- Х-сцепленному доминантному;
- Х-сцепленному рецессивному.
В связи с этим, классифицируется очень много разных форм СМА.
Детская форма СМА наследуется по аутосомно-рецессивному типу: если оба родителя — носители, то заболеет четвертая часть их потомства.
Аутосомно-доминантный тип наследования СМА приводит к проявлению болезни у детей с вероятностью 50%, даже если болен всего один родитель.
Подробнее о сути наследования можно прочитать в статье о синдроме Марфана.
Виды спинальной амиотрофии
Спинальная мышечная атрофия разделяется на четыре формы:
- Младенческая (I) — спинальная мышечная атрофия Верднига-Гофмана: диагностируется с момента рождения до полугода.
- Промежуточная (II) — болезнь Дубовица: от семи месяцев до полутора лет.
- Юношеская (III) — б. Кюгельберга-Веландера: после полутора лет.
- Взрослая (IV): после 35 лет.
Симптомы спинально-мышечной атрофии
Общие симптомы при СМА:
- поражение проксимальных (средних) мышц и фасций;
- сохранение чувствительности в большинстве клинических случаев;
- задержки умственного и психического развития при спинальной мышечной амиотрофии крайне редки;
- возможна при некоторых видах атрофия не только мышц конечностей, но респираторных, жевательных и глотательных.
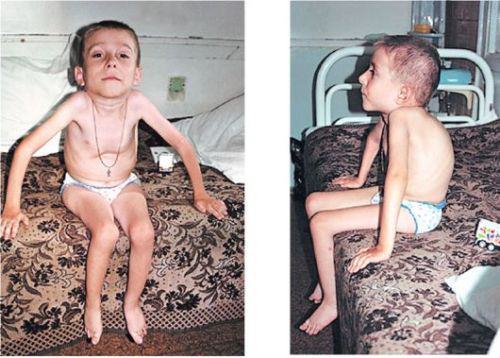
Степени тяжести СМА
- Самой тяжелой и неблагоприятной считается СМА первого вида (младенческая) — спинальная амиотрофия Верднига — Гоффмана, при которой дети не способны совершать активные движения, держать голову, самостоятельно сидеть. С большим трудом дается младенцу кормление, так как ему трудно сосать молоко и глотать его.
- Меньшей злокачественностью обладает болезнь Дубовица (II промежуточная форма СМА): при ней дети способны сидеть, держать голову и есть, однако ходить всё же неспособны.
- Наименее тяжелой является юношеская форма: несмотря на мышечную слабость, ребенок способен научиться ходить, однако заболевание хоть и медленно, но прогрессирует и может привести к ранней инвалидности.
- Четвертая взрослая форма СМА может привести из-за слабости м — ц проксимальных отделов к невозможности самостоятельного передвижения, выпадению рефлексов, но прогноз в отношение длительности жизни при этом остается благоприятным.

Другие виды спинально-мышечной атрофии
Кроме мутации в генах, вызывающих поражение проксимальных мышц, существуют подобные патологии, с разным типом наследования, приводящие к атрофии мышц и фасций дистальных (концевых отделов).
Список их довольно велик, сведем болезни в небольшую таблицу:
| Название СМА | Тип наследования | Особенности и симптомы |
| SMAX1 | Х -сцепленный рецессивный | Наблюдается в основном у пожилых, поражает бульбарные нервы черепа, вызывает нисходящий паралич. |
| SMAХ2 | Х — сцепл. рецессивный | Врожденная агрессивная форма, приводящая к смерти до 3 — х мес. Вызывает слабость, арефлексию, контрактуры и переломы. |
| SMAX3 | Х — сцепл. рецессивный | Поражает в основном мальчиков. Атрофия всех дистальных мышц. Медленное нарастание симптомов |
| Дистальная ДСМА1 | аутосомно — рецессивный | Врожденная, поражаются в основном руки, возможны тяжелые дыхательные нарушения |
| Дистальные формы ДСМА2 — ДСМА5 | аутосомно — рецессивный | Все четыре формы отличаются медленным прогрессированием, ДСМА5 диагностируется у молодых. |
| Дистальная СМА двух типов: VA и VB (DSMAVA и DSMAVB) | аутосомно-доминантный | Преимущественно атрофированы верхние конечности. |
| ДСМА тип 2D | аутосомно — рецессивный | Юношеское и взрослое заболевание с медленным развитием: поражаются и проксимальные, и дистальные м — цы вначале в голенях, затем в руках. |
| ДСМА тип 7А | аутосомно-доминантный | Очень редкая взрослая форма с поражением голосовых связок. |
| ДСМА тип 2А | аутосомно-доминантный | Разновидность болезни Шарко (аллельный тип) |
| Ювенильная SMA (тип HMN1) | аутосомно-доминантный | Встречается в юности |
| Врожденная спинальная амиотрофия | аутосомно-доминантный | Нарушение иннервации и атрофия м — ц бедер, стоп, коленей с контрактурой и деформацией; иногда поражается голосовые связки. |
| SMA Финкеля | аутосомно-доминантный | Начинается преимущественно в 35 — 37 лет, но зафиксированы случаи заболевания и в детском возрасте. Медленно развивается вначале в ногах, а затем в руках. Активность и рефлексы снижены, наблюдается непроизвольное дрожание (фасцикуляция). |
| SMA Джокела | аутосомно-доминантный | Поражаются у взрослых прокс. и дистальные м — цы. |
| SMA (тип LED1) | аутосомно-доминантный | Атрофия нижних конечностей у новорожденных. |
| SMA типа РМЕ | аутосомно — рецессивный | Атрофия дистальных мышц с нарушением иннервации и эпилептическими припадками |
| СМА с врожденными костными переломами | аутосомно — рецессивный | Тяжелые симптомы, как при болезни. Верднига-Гоффмана, отягощенные переломами. |
| СМА с гипоплазией | аутосомно-доминантный | Врожденная аномалия головного мозга с церебральными симптомами, микроцефалией и задержкой развития. |
| СМА ювенильная асимметричного типа | -------------- | Ею болеют молодые индийские мужчины |

В этой таблице следует обратить внимание на последние два вида спинальной амиотрофии:
- СМА с гипоплазией сопровождается отклонениями умственного и психического развития, что не характерно для остальных видом болезни.
- Асимметричная ювенильная (индийская) амиотрофия не передается по наследству. При этом заболевание после двух-пяти лет вялого течения может стабилизироваться. Симптомы фасцикуляции при этой специфической форме наблюдаются редко.
Лечение спинальной амиотрофии
Вылечить подобного рода болезни, как и любую наследственную патологию, затрагивающую спинной или головной мозг, кардинально невозможно. Эффективность многих используемых сегодня лекарств при терапии амиотрофии не доказана. Суть лечения сводится к увеличению белка, участвующего в формировании двигательных нейронов SMN.
Способы лечения СМА
Так, в основном применяются следующие препараты:
- вальпроевая кислота;
- оксибутират натрия;
- нусинерсен (новое лекарство, введенное в США в 2016 г. для лечения СМА).
Также необходимы поддерживающее лечение (ЛФК, массаж, физиотерапия), специальная белковая диета, при которой учитываются возможные противопоказания. Больные, лишенные возможности самостоятельного передвижения, нуждаются в социальной опеке.

Профилактика СМА
Профилактика спинальной амиотрофии возможна лишь в плане раннего выявления мутации в эмбриональном периоде: проводится ДНК-анализ при помощи специальной биопсии. Положительный результат анализа может быть основанием для прерывания беременности, если так решит мать.